|
Measuring multiple molecular types in the blood plasma from persons can give information about their health. If we measure them in large numbers (ideally the presence or absence of all of that are possible!) then we talk about proteomics, lipidomics and metabolomics for proteins, lipids and metabolites in general. These molecules are inter-related. For example, proteins synthesize and degrade lipids and bind metabolites. If we have such multiple omics datasets from the same person (and time point) we can better interpret the data in terms of pathways and processes activated, which then can tell us about how a disease is progressing or if a therapy is working. We provide methods to integrate these data in works like this one: Anyaegbunam, A.U., A. Vagiona, V. ten Cate, K. Bauer, T. Schmidlin, U. Distler, S. Tenzer, E. Araldi, L. Bindila, P. Wild and M.A. Andrade-Navarro. 2025. A map of the lipid-metabolite-protein network to aid multi-omics integration. Biomolecules. 15, 484. PubMed: 40305217 |

|
The diagram on the left represents a selection of feed-forward loops (FFLs) from a miRNA, a transcription factor and a gene they target. The scores are based on expression of FFL members in scRNA samples. The matrix on the right describes how the fraction of these genes (rows) that expresses alternative polyadenylation (APA) for transcripts including binding sites for a specific miRNA changes from cell to cell (columns). Combining this information, we can select APA patterns that are likely to be relevant for disease, for example, present in cancer cells that escape regulation by particular miRNAs. This approach was used here: Cihan, M., M. Sprang, G. Schmauck and M.A. Andrade-Navarro. 2025. Unveiling cell-type-specific microRNA networks through alternative polyadenylation in glioblastoma. BMC Biology. 23, 15. PubMed: 39838429 |

|
The study of homorepeats (or polyX), stretches of consecutive repeated amino acids in protein sequences has been a line of work in our group for a while. They are present in most human proteins and they are even more frequent in species like Plasmodium falciparum. We develop methods for their characterization and we predict their function by context. Everything started with polyQ (see the figure), which emerges in protein families with many protein interactions, tends to be inserted in a disordered region following an alpha helix, and often is followed by a P-rich region emerging C-terminal to the polyQ. Homorepeats have properties of low complexity regions and of repeats and the rules governing their structure and evolution are very different from those of globular domains. We explain and use these concepts in these papers: Mier, P. and M.A. Andrade-Navarro. 2021. Between interactions and aggregates: the polyQ balance. Genome Biology and Evolution. 13, evab246. PubMed: 34791220 Mier, P., C. Elena-Real, J. Cortés, P. Bernadó and M.A. Andrade-Navarro. 2022. The sequence context in poly-Alanine regions: Structure, function and conservation. Bioinformatics. 38, 4851-4858. PubMed: 36106994 |
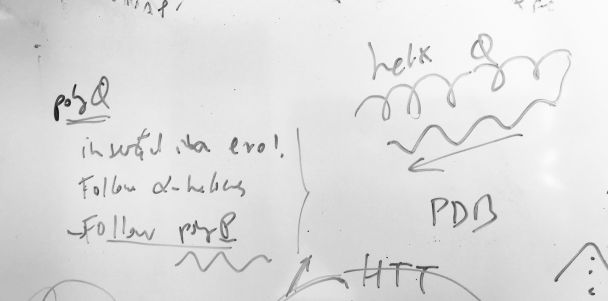
|
The sequencing of thousands of genomes from species of different taxa gives us unprecedented possibilities to evaluate the biological relevance of features in protein sequences that are apparently non-structured, such as low complexity regions. The diagram indicates a comparison across three species (Sp1, Sp2 and Sp3) focused on rows of orthologous proteins (lines, Core proteome) some of which have a feature (ball) and some that do not. Comparisons focused strictly on orthologs allow finding trends about the types of features, proteins that have them and positions in the sequences, which we use to make interpretations about the function of these features. You can see how we have used this approach in these papers: Kastano, K., G. Erdős, P. Mier, G. Alanis-Lobato, V.J. Promponas, Z. Dosztányi and M.A. Andrade-Navarro. 2020. Evolutionary study of disorder in protein sequences. Biomolecules. 10, 1413. PubMed: 33036302 Mier, P. and M.A. Andrade-Navarro. 2021. The conservation of low complexity regions in bacterial proteins depends on the pathogenicity of the strain and subcellular location of the protein. Genes. 12, 451. PubMed: 33809982 |

|
IT (Information Technology) groups in universities and research centers all over the world are key to the maintenance of computational services including communication and security. In our case, the JGU IT group performs additional tasks that are crucial to our research, maintaining a server where our web tools run, disk space where we store terabytes of data with archiving and backups, and computer clusters where we run our heavier calculations. For this reason, we really try hard not to give them unnecessary work, but sometimes we do make mistakes. That is why a sudden email from the IT group after submitting a very large computational task could make us jump in the chair. |
 by anonymous group leader
by anonymous group leader
|
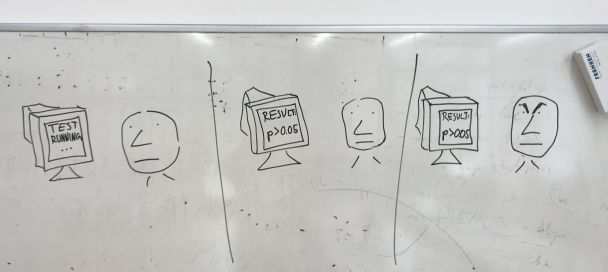
As part of our routine work we often obtain results by analysis of biological data that need to be assessed for significance with a statistical test. For example, say that we observe that a percentage of human genes related to a particular function, e.g. kinases, have a DNA motif in their promoters, e.g. GATTACA. If this DNA motif occurs very often by chance in the genome then the result may not be signicant: it could due to chance. We might test this by randomizing the genome and counting again how many times the DNA motif GATTACA appears in front of kinase encoding genes: this is our null-hypothesis. If we repeat this many times, the fraction of times we observe that the motif is equally or more frequent becomes a P-value that indicates the probability of the observation being due to chance. That's why we need to simulate the null-hypothesis in the computer and wait. And if the p-value is > 0.005 we are unhappy. |
 by anonymous master student
by anonymous master student
|
In this diagram each dot or plus sign represents a sample of cells, hES for human embryonic stem cells, iPS for induced pluripotent stem cells, Fib for fibroblasts. The position of the samples in the graph indicates in a simplified way how similar they are regarding their gene expression: close points have similar gene expression. Here, we would see that the iPS samples would have similar gene expression to hES, which would fit a reprogramming goal. However, the three samples in the box with the arrow would be somehow the result of fibroblasts that reprogrammed half way to iPS. Further efforts would be needed to understand which went wrong and what can be optimized. We are looking for genes and chemicals allowing us to reprogram cells from one type to another in a test-tube. You can read more about this in a related publication: Cheng, X., H. Yoshida, D. Raoofi, S. Saleh, H. Alborzinia, F. Wenke, A. Göhring, S. Reuter, N. Mah, H. Fuchs, M.A. Andrade-Navarro, J. Adjaye, S. Gul, J. Utikal, R. Mrowka and S. Wölfl. 2015. Ethyl 2-((4-chlorophenyl)amino)thiazole-4-carboxylate and derivatives are potent inducers of Oct3/4. J. Med. Chem. 58, 5742-5750. PubMed: 26143659 |

|
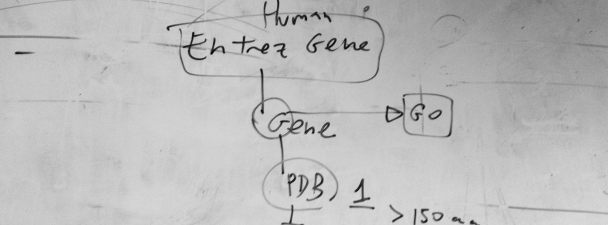
This is a draft of a flow chart for the selection of proteins to train a Support Vector Machine. The appropriate choice of examples for training and benchmark is a very important initial step in the development of methods for prediction in computational biology. In this case, we were working on a method to detect protein subcellular location based on amino acid composition and exposure. Therefore, we thought to start with all human protein sequences associated to genes in the Entrez NCBI database, then to take one associated to each human gene, for which we can obtain functional (GO) information, including subcellular location, and then to take those whose structure is deposited in the PDB database, provided that the solved structure had more than 150 amino acids. You can read the rest of the story in the related publication: Mer, A.S. and M.A. Andrade-Navarro. 2013. A novel approach for protein subcellular location prediction using amino acid exposure. BMC Bioinformatics. 14, 342. [NYCE] PubMed: 24283794 |